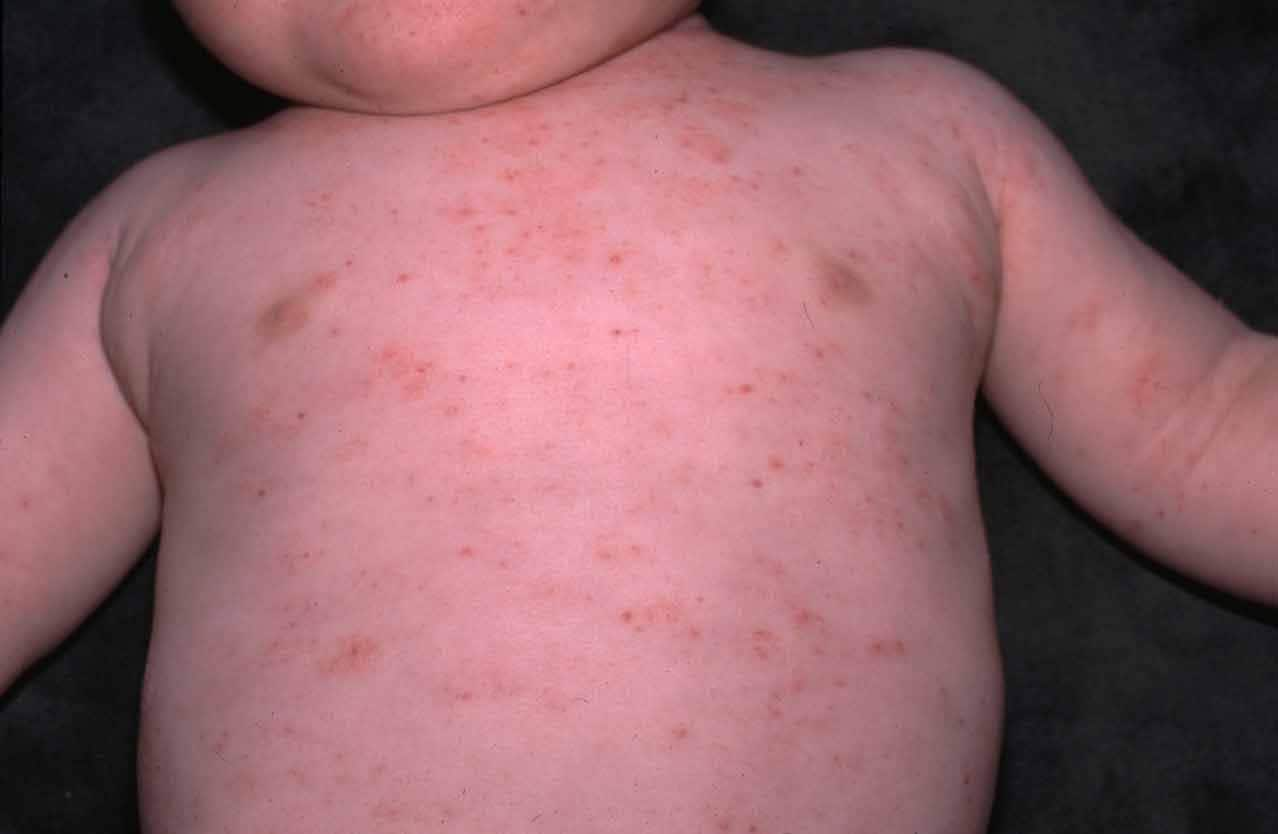
 El síndrome de Wiskott-Aldrich
ha sido tildado de “curioso” y “enigmático” y “desconcertante”debido a la
dificultad que existe para poder explicar sus defectos. Se trata de una
enfermedad recesiva ligada al cromosoma X que se carácteriza por
trombocitopenia (disminución de la cantidad de plaquetas), eccema y una
importante tendencia al desarrollo de infecciones de repetición que provocan la
muerte precoz del paciente. (Cotran, 2000)
El síndrome de Wiskott-Aldrich
ha sido tildado de “curioso” y “enigmático” y “desconcertante”debido a la
dificultad que existe para poder explicar sus defectos. Se trata de una
enfermedad recesiva ligada al cromosoma X que se carácteriza por
trombocitopenia (disminución de la cantidad de plaquetas), eccema y una
importante tendencia al desarrollo de infecciones de repetición que provocan la
muerte precoz del paciente. (Cotran, 2000)
Patogenia
El timo es morfológicamente
normal, al menos en las primeras fases de la enfermedad, pero progresivamente
se va produciendo una disminución de los linfocitos T de la sangre periférica y
de las áreas paracorticales (dependientes del timo) de los gánglios linfáticos,
con pérdida variable de la inmunidad celular. Los pacientes no fabrican
anticuerpos frente a antígenos de tipo polisacárido y su respuesta a los
antígenos proteicos es escasa. Los niveles séricos de IgM son bajos, pero los
de IgG suelen ser normales. Paradójicamente, los niveles de IgA y IgE suelen
estar elevados. Los pacientes tienden a desarrollar linfomas malignos. El
defecto genético de la enfermedad se encuentra en Xp11.23, lugar donde se
encuentra el gen de la enfermedad, llamado proteína del síndrome de
Wiskott-Aldrich. Esta proteína multifuncional interviene en el mantenimiento de
la integridad del citoesqueleto y en la transducción de señales. No se conoce
cual es la responsabilidad en la función normal de linfocitos y plaquetas. El
único tratamiento es el transplante de médula ósea. (Cotran, 2000)
Herencia
 El SWA se hereda como un
trastorno recesivo ligado al cromosoma X. Sólo los varones se ven afectados por
esta enfermedad. Dado que esta es una enfermedad heredada transmitida como un
rasgo recesivo ligado al cromosoma X, podría haber hermanos tíos maternos (el
hermano de la madre del paciente) con síntomas similares. Es posible que la
historia familiar sea totalmente negativa con respecto a la enfermedad debido
al tamaño reducido de la familia o a que se ha producido una nueva mutación. Se
cree que alrededor de una tercera parte de los casos de pacientes con SWA
recién diagnosticados se deben a una nueva mutación ocurrida en el momento de
la concepción. (inmunodeficiency
foundation, 2007)
El SWA se hereda como un
trastorno recesivo ligado al cromosoma X. Sólo los varones se ven afectados por
esta enfermedad. Dado que esta es una enfermedad heredada transmitida como un
rasgo recesivo ligado al cromosoma X, podría haber hermanos tíos maternos (el
hermano de la madre del paciente) con síntomas similares. Es posible que la
historia familiar sea totalmente negativa con respecto a la enfermedad debido
al tamaño reducido de la familia o a que se ha producido una nueva mutación. Se
cree que alrededor de una tercera parte de los casos de pacientes con SWA
recién diagnosticados se deben a una nueva mutación ocurrida en el momento de
la concepción. (inmunodeficiency
foundation, 2007)
Diagnóstico
El
síndrome debe sospecharse en varones con
sangrado asociado con eccema severo o moderado. El diagnóstico se corrobora con
exámenes de laboratorio; la biometría hemática confirma plaquetopenia y
plaquetas pequeñas. Por citometría de flujo el número de linfocitos circulantes
puede ser normal, estar discreta o francamente disminuido a expensas de
linfocitos T; los linfocitos B pueden ser normales o estar moderadamente disminuidos
.Las concentraciones de las IgG se encuentran frecuentemente en rango normal;
la IgM está disminuida y las inmunoglobulinas IgA e IgE se encuentran elevadas.
El diagnóstico definitivo es detectar la mutación en el gen WASP (Román Jiménez, 2010)
Pronóstico
Hace tres décadas, el Síndrome
de Wiskott-Aldrich clásico era una de las inmunodeficiencias primarias más
graves, cuya esperanza de vida era sólo de
entre 2 y 3 años. Aunque sigue siendo una enfermedad grave con
complicaciones que pueden poner la vida
en peligro, muchos varones afectados pasan la
pubertad, llegan a la edad adulta, llevan vidas productivas y tienen sus
propias familias. Los pacientes de más
edad sometidos a trasplantes de médula ósea
ya tienen hoy en día entre veinte y treinta años, y parecen estar
curados, pues no han desarrollado tumores malignos ni enfermedades autoinmunes.
(inmunodeficiency foundation, 2007)
No hay comentarios:
Publicar un comentario